


Mcapitata Development 16S Analysis Part 2
This post details QC and QIIME analysis for the 16S analysis adapted from the pipeline developed by Emma Strand.
Loading 16S Data in QIIME2
Log into Andromeda and navigate to data folder. If off campus, use VPN connection via AnyConnect.
ssh -l ashuffmyer ssh3.hac.uri.edu
cd /data/putnamlab/ashuffmyer/AH_MCAP_16S
6. Create metadata files
Before proceeding, read QIIME2 webpage and tutorials.
We need to create two metadata files: Sample manifest file and sample metadata file
Create a directory for metadata in the AH_MCAP_16S directory.
mkdir metadata
First, create a list of all samples and file paths to help create metadata files.
cd raw_data
find $PWD -type f ! -name filepath.csv > filepath.csv
find $PWD -type f ! -name filenames.csv ! -name filepath.csv -printf "%P\n" >filenames.csv
mv filenames.csv /data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata
mv filepath.csv /data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata
Outside of Andromeda, move these files to your local computer.
scp ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata/filenames.csv ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/16S
scp ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata/filepath.csv ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/16S
Sample manifest file
Create a sample manifest file with the following columns: sample-id, forward-absolute-path, and reverse-absolute-path. Note that this is different from previous pipeline versions. I originally create a file with a column for direction (e.g., forward or reverse) and had errors loading data due to two lines in the manifest per sample.
Sample manifest file should have:
sample-id (e.g., WSH201)
forward-absolute-path (file path of R1 read)
reverse-absolute-path (file path of R2 read)
Save as a tab-delimited file .txt.
This file is named sample_manifest.txt.
Copy back into Andromeda.
scp ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/16S/sample_manifest.txt ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata/
The file looks like this:
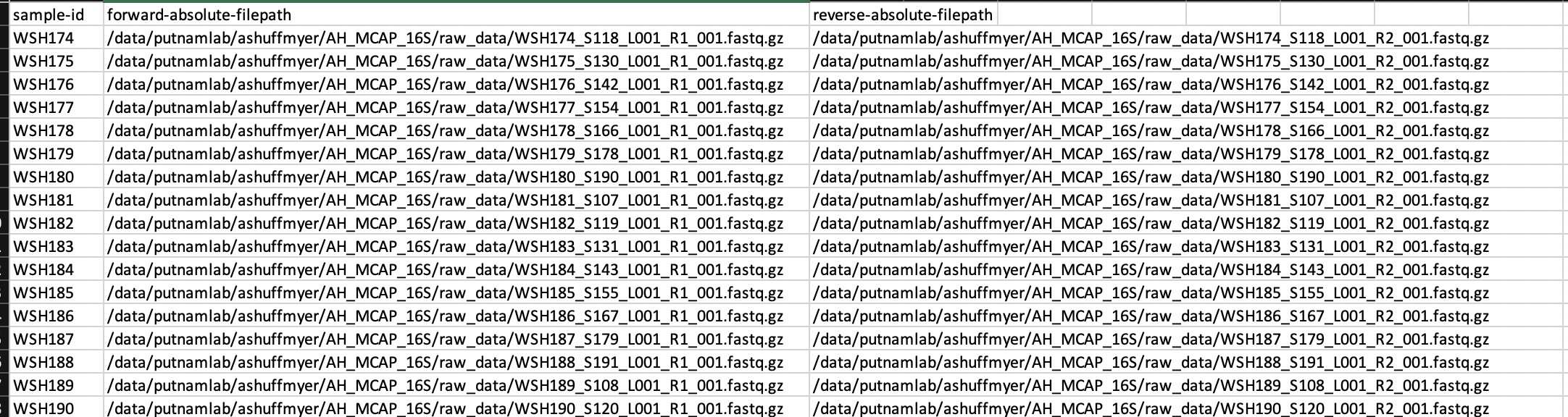
Sample metadata file
Create a sample metadata file based on QIIME2 requirements (see links above). Metadata includes the first row as a header and the second row as the data type (#q2:types) with metadata starting in the third row. Save this file as a tab-delimited txt file as for the manifest file.
Platemaps with sample names spreadsheet here.
I created this file manually on my computer and then copied into Andromeda.
scp ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/16S/sample_metadata.txt ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata/
The file looks like this:
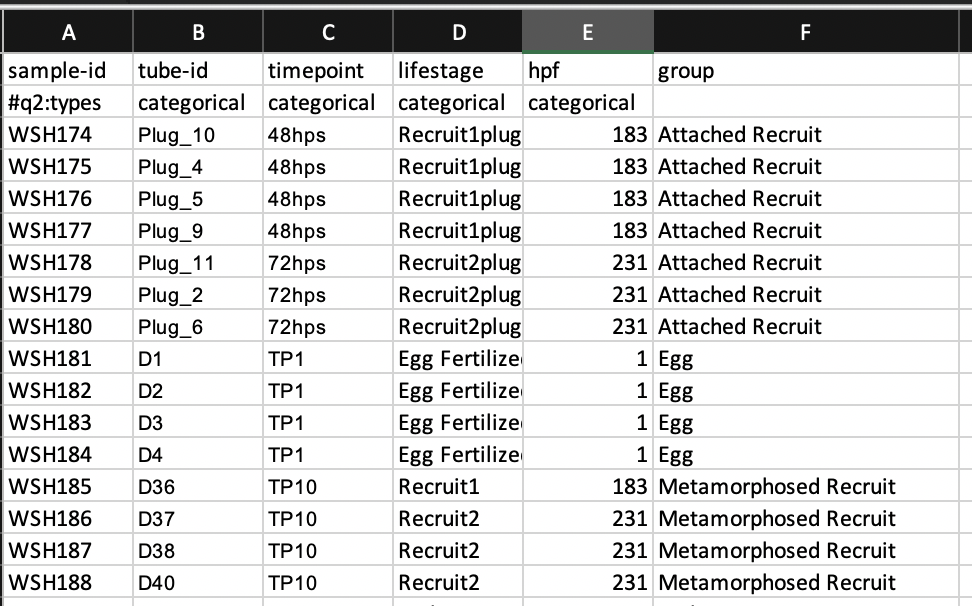
7. Input data into QIIME2
Next, input sample data into QIIME2. The settings were copied from E. Strand’s pipeline for this preliminary analysis.
More information on importing data here.
- Sequence Data with Sequence Quality Information: because we have fastq files, not fasta files.
- FASTQ data in paired-end demultiplexed format: because our samples are already demultiplexed and we have 1 file per F and R.
- Input path directs to the sample manifest file created above.
- PairedEndFastqManifestPhred33 option requires a forward and reverse read. This assumes that the PHRED offset for positional quality scores is 33 - more info here.
Enter interactive mode and load modules.
interactive
module load Miniconda3/4.9.2
module load Python/3.8.2-GCCcore-9.3.0
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
module load cutadapt/2.10-GCCcore-9.3.0-Python-3.8.2
module load QIIME2/2021.8
Move the manifest file in with the data files.
mv /data/putnamlab/ashuffmyer/AH_MCAP_16S/metadata/sample_manifest.txt /data/putnamlab/ashuffmyer/AH_MCAP_16S/raw_data/
Run in the AH_MCAP_16S directory.
Note that I had to create a new conda environment, the previous environment was not present. This step is not needed if conda activate works.
conda create -n AH_MCAP_16S
conda activate AH_MCAP_16S
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path raw_data/sample_manifest.txt \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path AH-MCAP-16S-paired-end-sequences1.qza
Running this script I got the following error:
ValueError: numpy.ndarray size changed, may indicate binary incompatibility. Expected 88 from C header, got 80 from PyObject
It seems that we need to update conda/numpy so that it can be accessed in the cluster.
conda upgrade numpy may be the correct solution. Contacting Kevin Bryan to ask about the option to upgrade Conda and numpy.
K. Bryan updated QIIME2 to QIIME2021.8 on 20211229 this includes an upgrade to numpy.
Try to import data again. Must re activate conda environment and load modules in interactive mode each time you exit interactive mode.
interactive
module load Miniconda3/4.9.2
conda activate AH_MCAP_16S
module load QIIME2/2021.8
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path raw_data/sample_manifest.txt \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path AH-MCAP-16S-paired-end-sequences1.qza
Success! Data imported.
Output reads:
Imported raw_data/sample_manifest.txt as PairedEndFastqManifestPhred33V2 to AH-MCAP-16S-paired-end-sequences1.qza
Now the QIIME artifact named AH-MCAP-16S-paired-end-sequences1.qza lives in the AH_MCAP_16S directory.