


Mcapitata Development ITS2 Analysis Part 2
This post details SymPortal analysis for the ITS2 analysis pipeline modified from Kevin Wong pipeline.
ITS2 analysis in SymPortal
5. Prepare Symportal settings and files
More information on Symportal here.
The first step of analysing an ITS2 dataset is to load it into the SymPortal framework’s database. In this step, SymPortal will perform all quality control filtering of the sequence data and convert the raw sequence data into database objects. There is no need for pre filtering or trimming in this case.
Copy SymPortal into home directory for permission access.
Navigate to home directory and download SymPortal.
ssh -l ashuffmyer ssh3.hac.uri.edu
cd /data/putnamlab/ashuffmyer/
rsync -rl /opt/software/SymPortal/0.3.21-foss-2019b/ ./SymPortal/
This step takes a few minutes to complete. There is now a SymPortal directory.
Modify settings and config files.
cd SymPortal
module load SymPortal
mv settings_blank.py ./settings.py
mv sp_config_blank.py ./sp_config.py
base64 /dev/urandom | head -c50
The last command will display a secret key in the header. Copy this key. Open settings.py.
nano settings.py
Paste secret key in settings.py file in SECRET_KEY = ‘qxwykujthzogaKEyHZSFCjmZlTGrt/ureW3Ppw4wQdhPAuCeEn’ and save it.
Open the config file and edit username and email.
nano sp_config.py
user_name = "undefined" --> user_name = "ashuffmyer"
user_email = "undefined" --> user_email = "ashuffmyer@uri.edu"
Create the SymPortal conda environment
Next, create a conda environment to work in. You can do this in interactive mode, but there is a long install.
interactive
module load Miniconda3/4.9.2
module load SymPortal
conda env create -f $EBROOTSYMPORTAL/symportal_env.yml
exit
Download often failed, so downloaded in several sessions. It appears that it recognizes what has already been downloaded and doesn’t seem to repeat what was downloaded in a previous session.
Internet connection didn’t allow for the download easily. Trying to download in a bash script overnight.
nano install.sh
#!/bin/bash
#SBATCH --job-name="install"
#SBATCH -t 500:00:00
#SBATCH --mem=250GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=ashuffmyer@uri.edu
#SBATCH --exclusive
module load Miniconda3/4.9.2
module load SymPortal
conda env create -f $EBROOTSYMPORTAL/symportal_env.yml
echo "Success!" $(date)
sbatch install.sh
Ran overnight and completed, now I will try to run analyses to see if the downloads worked correctly.
I got these error codes for each package:
CondaVerificationError: The package for django located at /home/ashuffmyer/.con$
appears to be corrupted. The path 'site-packages/django/views/generic/base.py'
specified in the package manifest cannot be found.
However, when I tried to initiate download again it said that the environment already existed. Proceeding with analysis.
Create a reference database
Create a reference data base in our SymPortal environment.
cd SymPortal
interactive
module load Miniconda3/4.9.2
eval "$(conda shell.bash hook)"
conda activate symportal_env
module load SymPortal/0.3.21-foss-2020b
python3 manage.py migrate
##### Populating reference_sequences
python3 populate_db_ref_seqs.py
module unload SymPortal
exit
I had an issue with djano here (error loading django) when entering the python3 manage.py migrate function.
The issue is likely that it was trying to run the SymPortal within the data directory and it should be installed in the home ‘ashuffmyer’ directory.
Trying instead to run as a bash script using the conda paths from Kevin Wong’s scripts that direct SymPortal to the home directory.
cd scripts
nano reference.sh
#!/bin/bash
#SBATCH --job-name="SP_reference"
#SBATCH -t 500:00:00
#SBATCH --mem=120GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=ashuffmyer@uri.edu
#SBATCH -D /data/putnamlab/ashuffmyer/SymPortal
#SBATCH --exclusive
module load Miniconda3/4.9.2
eval "$(conda shell.bash hook)"
conda activate symportal_env
module load SymPortal/0.3.21-foss-2020b
module unload
export PYTHONPATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/lib/python3.7/site-packages:$PYTHONPATH
export PATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/bin:$PATH
python3 manage.py migrate
python3 populate_db_ref_seqs.py
module unload SymPortal/0.3.21-foss-2020b
echo "Mission Complete!" $(date)
This works as a script! The export paths seem to fix the problem.
Test installation and reference database
SymPortal was not built to run on a cluster, so permission access was tricky as you cannot write into the master installation module. Therefore, we need to change the python and SymPortal paths to the ones we created in our own directory. Additionally, some of the dependencies needed by SymPortal are only in the master installation module. To use these dependencies but write into our own SymPortal framework, we must load then unload the master SymPortal module in our script.
cd scripts
nano symportal_setup.sh
#!/bin/bash
#SBATCH --job-name="SP_setup"
#SBATCH -t 500:00:00
#SBATCH --mem=120GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=ashuffmyer@uri.edu
#SBATCH -D /data/putnamlab/ashuffmyer/SymPortal
#SBATCH --exclusive
module load Miniconda3/4.9.2
eval "$(conda shell.bash hook)"
conda activate symportal_env
module load SymPortal/0.3.21-foss-2020b
module unload
export PYTHONPATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/lib/python3.7/site-packages:$PYTHONPATH
export PATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/bin:$PATH
##### Checking installation
python3 -m tests.tests
echo "Mission Complete!" $(date)
Job completed.
6. Load experimental data
Create metadata file
First, create a metadata file from this template from SymPortal.
Export a list of file names that will be manually added to this sheet for file names for forward and reverse files.
cd raw_data
find $PWD -type f ! -name filenames.csv ! -name filepath.csv -printf "%P\n" >filenames.csv
mv filenames.csv ../metadata/
Outside of Andromeda:
scp ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_ITS2/metadata/filenames.csv ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/ITS2
Manually create the metadata file then add back to Andromeda as a .csv of the first page of the template.
Platemaps with sample names spreadsheet here.
mkdir metadata
Outside of Andromeda:
scp ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/ITS2/AH_MCAP_ITS2_meta.csv ashuffmyer@bluewaves.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_ITS2/metadata/
Loading the data
Create a script in the scripts directory.
cd ../scripts
nano symportal_load.sh
#!/bin/bash
#SBATCH --job-name="SP_load"
#SBATCH -t 500:00:00
#SBATCH --mem=120GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=ashuffmyer@uri.edu
#SBATCH -D /data/putnamlab/ashuffmyer/SymPortal
#SBATCH --exclusive
module load Miniconda3/4.9.2
eval "$(conda shell.bash hook)"
conda activate symportal_env
module load SymPortal/0.3.21-foss-2020b
module unload
export PYTHONPATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/lib/python3.7/site-packages:$PYTHONPATH
export PATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/bin:$PATH
main.py --load /data/putnamlab/ashuffmyer/AH_MCAP_ITS2/raw_data \
--name Mcap_Development \
--num_proc $SLURM_CPUS_ON_NODE \
--data_sheet /data/putnamlab/ashuffmyer/AH_MCAP_ITS2/metadata/AH_MCAP_ITS2_meta.csv
Script output indicates 0 sequences “thrown out” due to being too divergent from references. Output file will have information on number of sequences and QC steps.
7. Run analysis in SymPortal
Create a script within the scripts directory.
nano sp_analysis.sh
#!/bin/bash
#SBATCH --job-name="SP_analysis"
#SBATCH -t 500:00:00
#SBATCH --mem=120GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --mail-type=BEGIN,END,FAIL
#SBATCH --mail-user=ashuffmyer@uri.edu
#SBATCH -D /data/putnamlab/ashuffmyer/SymPortal
#SBATCH --exclusive
module load Miniconda3/4.9.2
eval "$(conda shell.bash hook)"
conda activate symportal_env
module load SymPortal/0.3.21-foss-2020b
module unload
export PYTHONPATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/lib/python3.7/site-packages:$PYTHONPATH
export PATH=/data/putnamlab/ashuffmyer/SymPortal/:/data/putnamlab/ashuffmyer/SymPortal/bin:$PATH
# Checking dataset number
./main.py --display_data_sets
# Running analysis
./main.py --analyse 5 --name MCAPdevelop_analysis --num_proc $SLURM_CPUS_ON_NODE
# Checking data analysis instances
./main.py --display_analyses
At first this script did not work, so I ran the script only with the #checking dataset number part first. In the slurm output file, this showed that I had 5 datasets. I then replaced the number in the #running analysis step with 5 and the script then worked. Then I ran the whole script again for the analysis.
8. Export data
Export the data to your computer.
Save and transfer the output files.
All data can be found in GitHub here.
scp -r ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/SymPortal/outputs ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/ITS2
9. Explore data
View the type_abundance_stacked_bar_plot.png and other .png files to view results. In this study, we saw a mix of C and D across life stages, which is expected for M. capitata. We can now do further exploration in R.
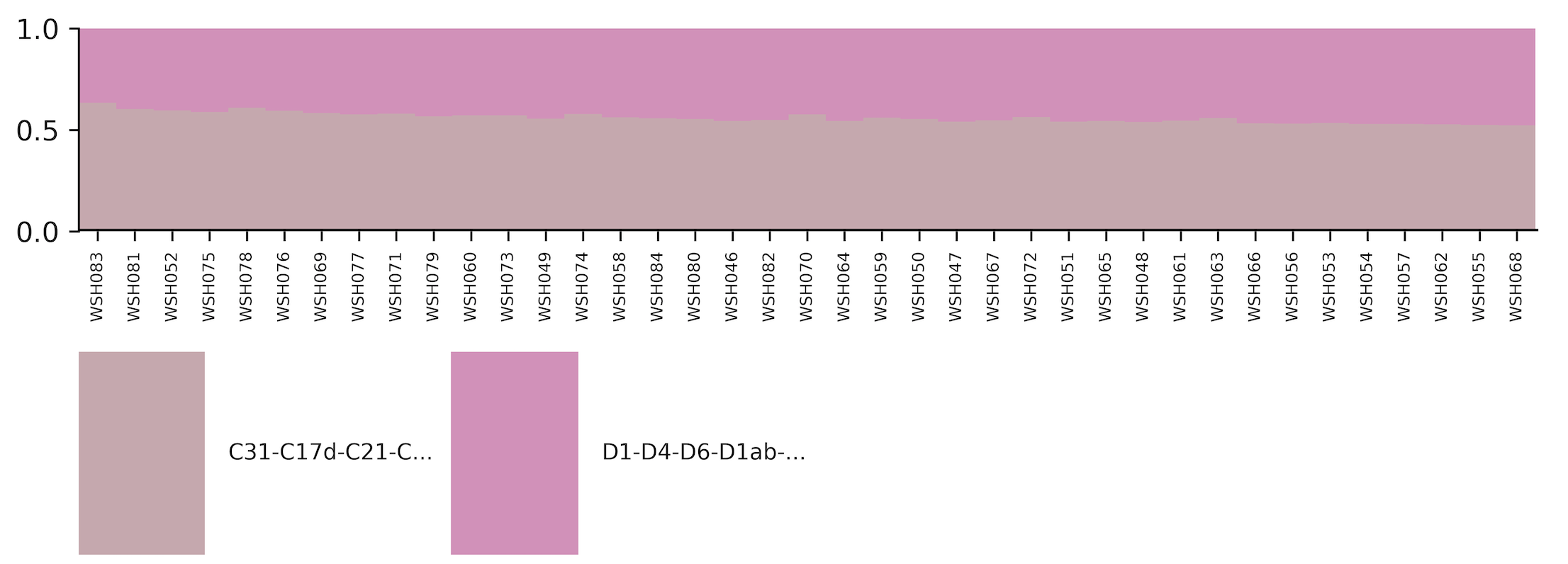
Next run the data in R to view profiles ordered by lifestage. Script located in the ITS2.Rmd file here.
ITS2 clade C = C31 ITS2 clade D = D1, associated species = S. glynnii and S. trenchii
Relative abundance by samples:
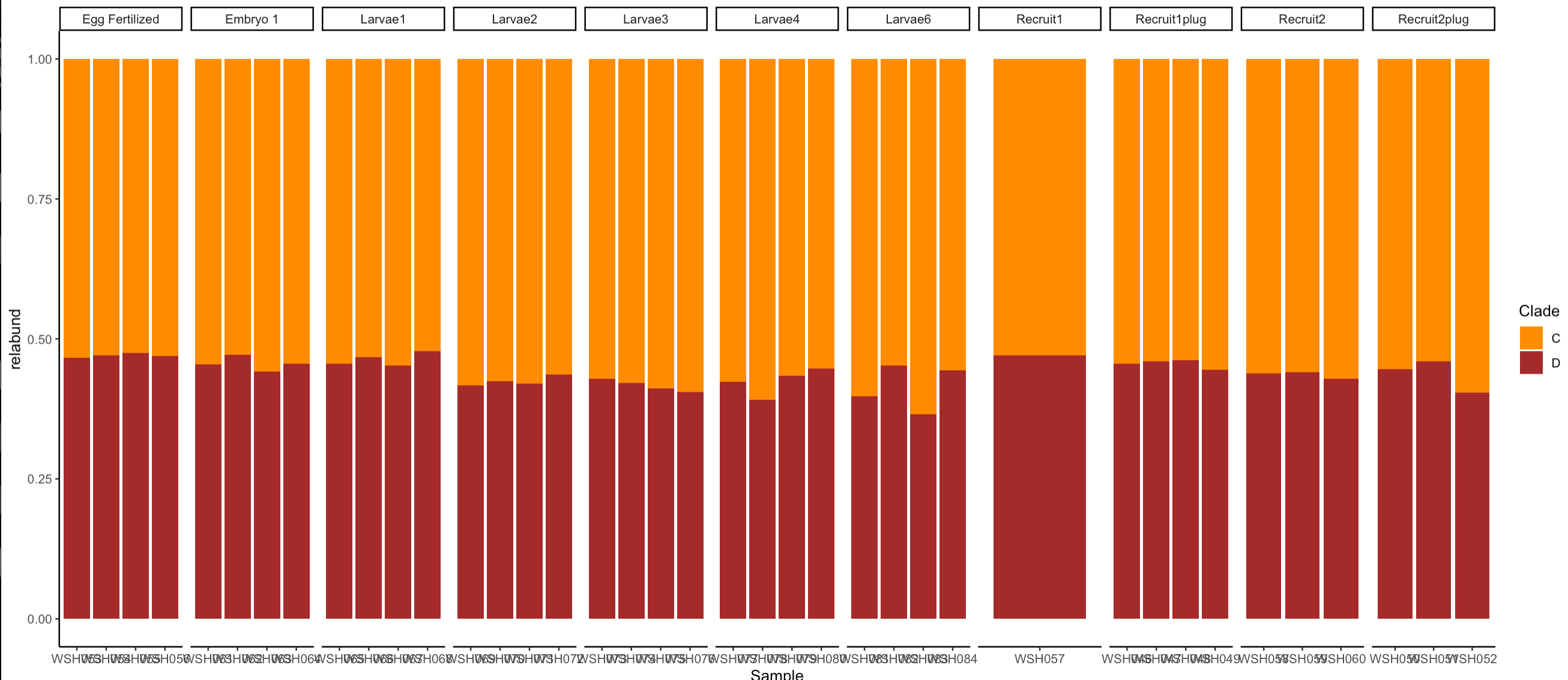
Relative abundance by lifestage:
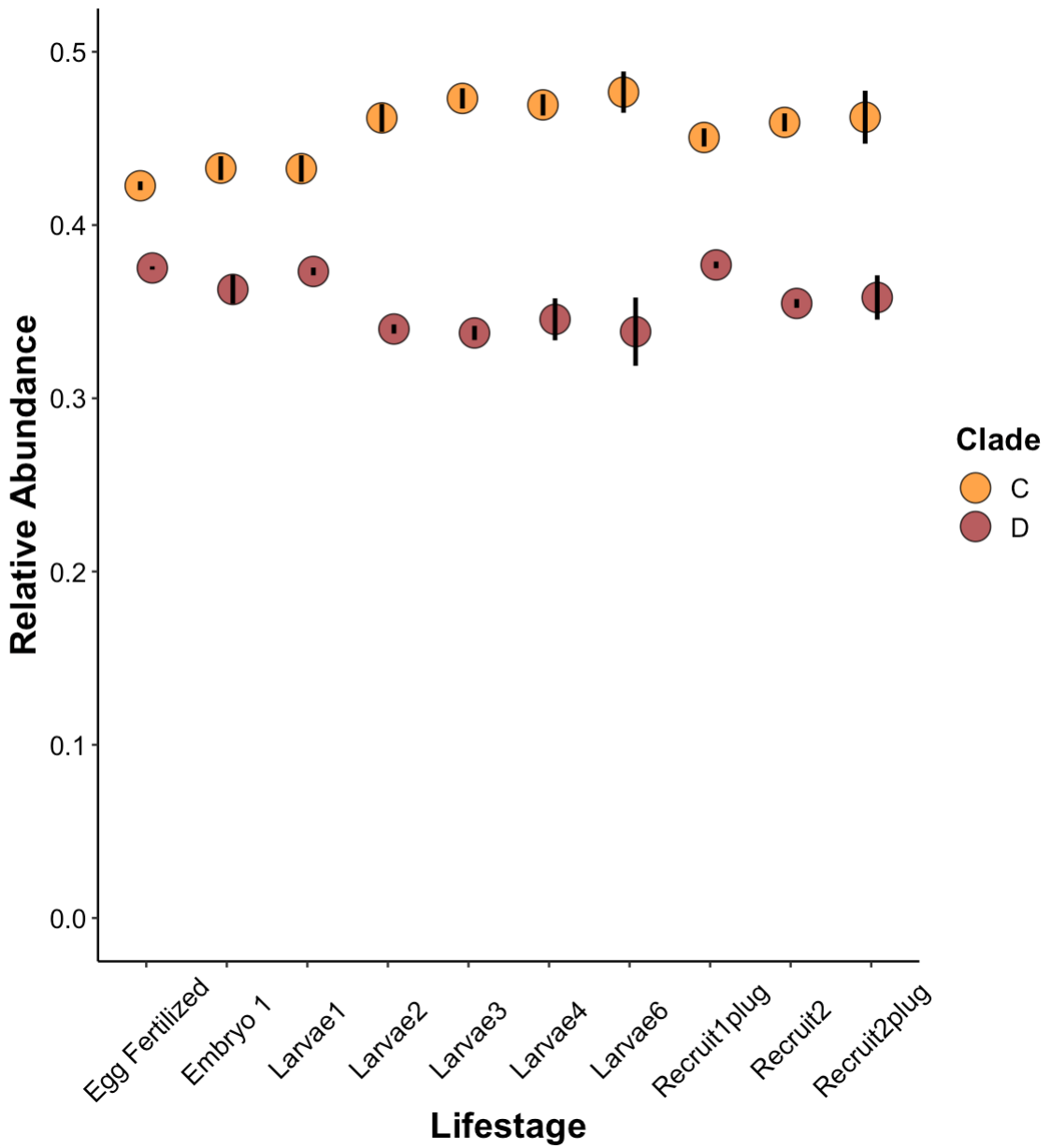
Anova analyses indicate significant differences in relative abundance of C and D between lifestages. It appears that C increases during development and D decreases.
Finally, move sequences to personal computer.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/AH_MCAP_ITS2/raw_data/*.gz ~/MyProjects/EarlyLifeHistory_Energetics/Mcap2020/Data/ITS2/Sequences