


QC of RNAseq files for Montipora capitata larval thermal tolerance 2023 project
This post details initial QC of the Montipora capitata 2023 larval thermal tolerance project RNAseq files. See my notebook posts and my GitHub repo for information on this project. Also see my notebook post on sequence download and SRA upload for these files.
1. Run MultiQC on raw data (untrimmed and unfiltered)
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq
cd scripts
nano qc_raw.sh
#!/bin/bash
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=20
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --output="%x_out.%j"
#SBATCH --error="%x_err.%j"
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences
# load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# fastqc of raw reads
fastqc *.fastq.gz
#generate multiqc report
multiqc ./ --filename multiqc_report_raw.html
echo "Raw MultiQC report generated." $(date)
Run the script.
sbatch qc_raw.sh
Run as Job ID 303829 on 23 Feb 2024
Finised by 24 Feb 2024.
Move .fastqc files to a QC folder to keep folder organized.
mkdir raw_fastqc
mv *fastqc.html raw_fastqc
mv *fastqc.zip raw_fastqc
mv multiqc* raw_fastqc
Download the multiQC report to my desktop to view (in a new terminal not logged into Andromeda).
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/raw_fastqc/multiqc_report_raw.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC
The raw sequence MultiQC report can be found on GitHub here.
Here are the things I noticed from the QC report:
See this previous post with statistics of samples provided by Azenta.
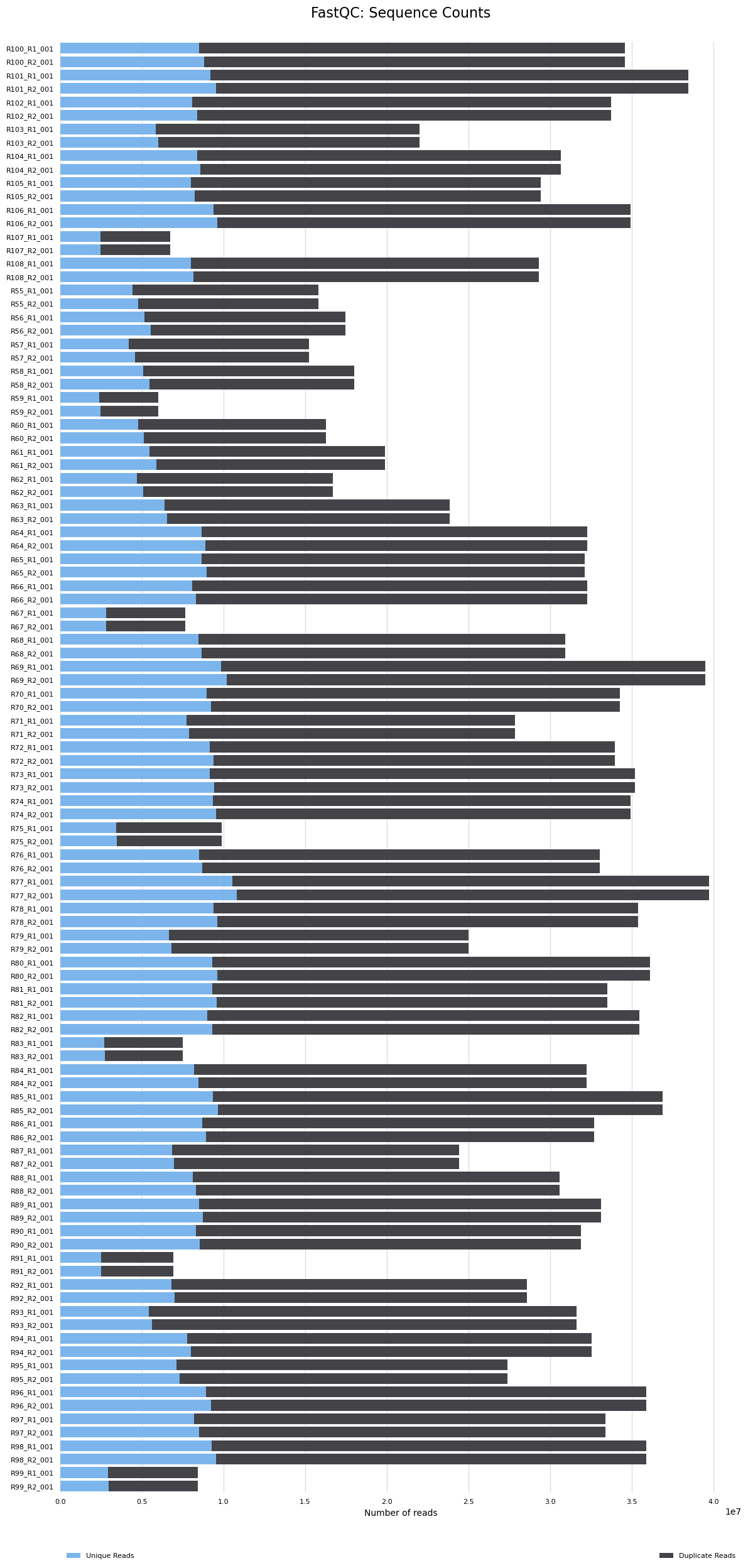
- Samples with lowest sequence counts
- Samples R75, R99, R67, R83, R91, R107, R59 have <10M sequence counts. These are randomly distributed between treatments, so if they are removed it doesn’t cause a problem. A standard approach is to have samples with >5M mapped reads for DEG analysis. If we have <5M reads after mapping they will likely be removed or we will need to see how or if results from these samples differ from others using PCA’s, etc.
-
I emailed Azenta on 20240226 to inquire about library prep QC, methods, and the option to resequence these low read depth libraries.
- R75: Bleached Cladocopium at 30°C; Batch 7; Qbit RNA 16.1; Nanodrop RNA 12.5
- R99: Nonbleached Mixed at 33°C; Batch 9; Qbit RNA 21.6; Nanodrop RNA 14.1
- R67: Wildtype at 27°C; Batch 8; Qbit RNA 20.7; Nanodrop RNA 18.6
- R83: Nonbleached Mixed at 33°C; Batch 7; Qbit RNA 18.15; Nanodrop RNA 13.5
- R91: Bleached Cladocopium at 33°C; Batch 7; Qbit RNA 25.2; Nanodrop RNA 18.6
- R107: Wildtype at 33°C; Batch 9; Qbit RNA 16.85; Nanodrop RNA 11.7
-
R59: Bleached Cladocopium at 27°C; Batch 9; Qbit RNA 17.75; Nanodrop RNA 10.7
- All other samples had RNA concentrations ranging form Qbit 15-40 and Nanodrop 9-25. They are not all from one batch either. Low sequence counts do not appear to correlate with original RNA extraction or concentration and may be due to library prep or something that happened during sequencing.
| Sample Name | % Dups | % GC | M Seqs |
|---|---|---|---|
| R77_R1_001 | 73.5% | 44% | 39.7 |
| R77_R2_001 | 72.9% | 44% | 39.7 |
| R69_R1_001 | 75.0% | 44% | 39.5 |
| R69_R2_001 | 74.2% | 44% | 39.5 |
| R101_R1_001 | 76.1% | 43% | 38.5 |
| R101_R2_001 | 75.2% | 44% | 38.5 |
| R85_R1_001 | 74.7% | 43% | 36.9 |
| R85_R2_001 | 73.8% | 44% | 36.9 |
| R80_R1_001 | 74.2% | 44% | 36.1 |
| R80_R2_001 | 73.4% | 44% | 36.1 |
| R96_R1_001 | 75.2% | 45% | 35.9 |
| R96_R2_001 | 74.3% | 45% | 35.9 |
| R98_R1_001 | 74.2% | 44% | 35.9 |
| R98_R2_001 | 73.4% | 44% | 35.9 |
| R82_R1_001 | 74.6% | 43% | 35.5 |
| R82_R2_001 | 73.8% | 44% | 35.5 |
| R78_R1_001 | 73.4% | 43% | 35.4 |
| R78_R2_001 | 72.8% | 44% | 35.4 |
| R73_R1_001 | 74.0% | 44% | 35.2 |
| R73_R2_001 | 73.3% | 44% | 35.2 |
| R106_R1_001 | 73.2% | 43% | 34.9 |
| R106_R2_001 | 72.4% | 44% | 34.9 |
| R74_R1_001 | 73.3% | 44% | 34.9 |
| R74_R2_001 | 72.7% | 44% | 34.9 |
| R100_R1_001 | 75.4% | 44% | 34.6 |
| R100_R2_001 | 74.5% | 44% | 34.6 |
| R70_R1_001 | 73.8% | 43% | 34.3 |
| R70_R2_001 | 73.0% | 44% | 34.3 |
| R72_R1_001 | 73.1% | 43% | 34.0 |
| R72_R2_001 | 72.3% | 44% | 34.0 |
| R102_R1_001 | 76.1% | 44% | 33.7 |
| R102_R2_001 | 75.1% | 44% | 33.7 |
| R81_R1_001 | 72.2% | 44% | 33.5 |
| R81_R2_001 | 71.4% | 45% | 33.5 |
| R97_R1_001 | 75.4% | 43% | 33.4 |
| R97_R2_001 | 74.6% | 44% | 33.4 |
| R89_R1_001 | 74.3% | 43% | 33.1 |
| R89_R2_001 | 73.6% | 44% | 33.1 |
| R76_R1_001 | 74.2% | 44% | 33.0 |
| R76_R2_001 | 73.7% | 44% | 33.0 |
| R86_R1_001 | 73.5% | 43% | 32.7 |
| R86_R2_001 | 72.7% | 44% | 32.7 |
| R94_R1_001 | 76.1% | 44% | 32.5 |
| R94_R2_001 | 75.5% | 44% | 32.5 |
| R64_R1_001 | 73.2% | 44% | 32.3 |
| R64_R2_001 | 72.5% | 44% | 32.3 |
| R66_R1_001 | 75.0% | 43% | 32.3 |
| R66_R2_001 | 74.3% | 44% | 32.3 |
| R84_R1_001 | 74.5% | 43% | 32.2 |
| R84_R2_001 | 73.7% | 44% | 32.2 |
| R65_R1_001 | 73.0% | 43% | 32.1 |
| R65_R2_001 | 72.1% | 44% | 32.1 |
| R90_R1_001 | 73.9% | 43% | 31.9 |
| R90_R2_001 | 73.2% | 44% | 31.9 |
| R93_R1_001 | 82.9% | 46% | 31.6 |
| R93_R2_001 | 82.3% | 46% | 31.6 |
| R68_R1_001 | 72.7% | 43% | 30.9 |
| R68_R2_001 | 72.1% | 44% | 30.9 |
| R104_R1_001 | 72.7% | 44% | 30.6 |
| R104_R2_001 | 72.0% | 44% | 30.6 |
| R88_R1_001 | 73.5% | 44% | 30.6 |
| R88_R2_001 | 72.8% | 45% | 30.6 |
| R105_R1_001 | 72.8% | 43% | 29.4 |
| R105_R2_001 | 72.1% | 44% | 29.4 |
| R108_R1_001 | 72.7% | 43% | 29.3 |
| R108_R2_001 | 72.2% | 44% | 29.3 |
| R92_R1_001 | 76.2% | 43% | 28.6 |
| R92_R2_001 | 75.5% | 44% | 28.6 |
| R71_R1_001 | 72.2% | 43% | 27.8 |
| R71_R2_001 | 71.7% | 44% | 27.8 |
| R95_R1_001 | 74.0% | 43% | 27.4 |
| R95_R2_001 | 73.3% | 44% | 27.4 |
| R79_R1_001 | 73.5% | 44% | 25.0 |
| R79_R2_001 | 72.7% | 45% | 25.0 |
| R87_R1_001 | 71.9% | 43% | 24.4 |
| R87_R2_001 | 71.5% | 43% | 24.4 |
| R63_R1_001 | 73.2% | 43% | 23.8 |
| R63_R2_001 | 72.6% | 44% | 23.8 |
| R103_R1_001 | 73.4% | 44% | 22.0 |
| R103_R2_001 | 72.7% | 44% | 22.0 |
| R61_R1_001 | 72.6% | 44% | 19.9 |
| R61_R2_001 | 70.4% | 44% | 19.9 |
| R58_R1_001 | 71.9% | 43% | 18.0 |
| R58_R2_001 | 69.6% | 44% | 18.0 |
| R56_R1_001 | 70.4% | 44% | 17.5 |
| R56_R2_001 | 68.3% | 45% | 17.5 |
| R62_R1_001 | 71.8% | 44% | 16.7 |
| R62_R2_001 | 69.5% | 45% | 16.7 |
| R60_R1_001 | 70.8% | 44% | 16.3 |
| R60_R2_001 | 68.6% | 45% | 16.3 |
| R55_R1_001 | 72.1% | 43% | 15.8 |
| R55_R2_001 | 69.8% | 44% | 15.8 |
| R57_R1_001 | 72.4% | 43% | 15.2 |
| R57_R2_001 | 70.1% | 44% | 15.2 |
| R75_R1_001 | 65.4% | 43% | 9.9 |
| R75_R2_001 | 65.1% | 44% | 9.9 |
| R99_R1_001 | 65.3% | 44% | 8.4 |
| R99_R2_001 | 64.9% | 44% | 8.4 |
| R67_R1_001 | 63.6% | 43% | 7.7 |
| R67_R2_001 | 63.2% | 44% | 7.7 |
| R83_R1_001 | 64.1% | 43% | 7.5 |
| R83_R2_001 | 63.5% | 44% | 7.5 |
| R91_R1_001 | 64.1% | 43% | 6.9 |
| R91_R2_001 | 63.7% | 44% | 6.9 |
| R107_R1_001 | 63.7% | 43% | 6.7 |
| R107_R2_001 | 63.2% | 44% | 6.7 |
| R59_R1_001 | 60.2% | 44% | 6.0 |
| R59_R2_001 | 59.2% | 44% | 6.0 |
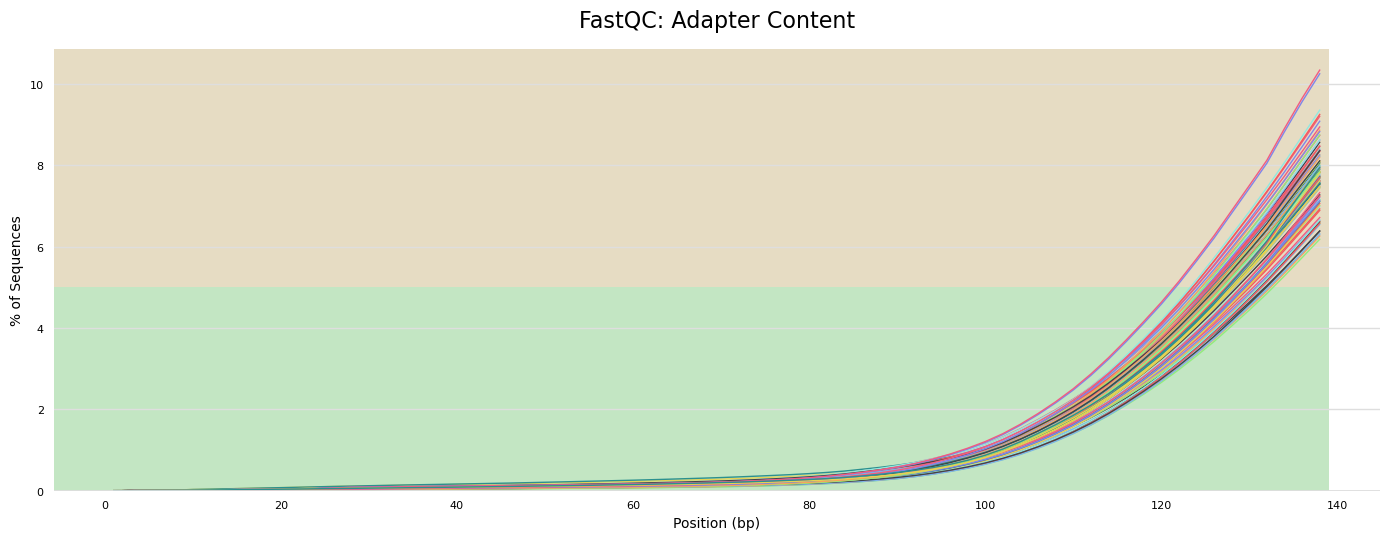
- Adapter content present in sequences, as expected, because we have not yet removed adapters.
- Some samples have warnings for GC content. We will revisit this after trimming.

- There are a high proportion of overrepresented sequences. This is expected with RNAseq data as there are likely genes that are highly and consistently expressed.

-
All reads are the same length (150 bp).
-
High quality scores.

- Low/no N content

2. Trimming adapters
I first ran a step to trim adapters from sequences. I will then generate another QC report to look at the results before making other trimming decisions.
I first moved the .md5 files to an md5 folder to keep only .fastq files in raw-sequences.
cd raw-sequences
mkdir md5_files
mv *.md5 md5_files
Make a new folder for trimmed sequences.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/
mkdir trimmed-sequences
cd scripts
nano trim_adapters.sh
Generate a script adapted from Jill Ashey’s script here.
I will use the following settings for trimming in fastp. Fastp documentation can be found here.
detect_adapter_for_pe \- This enables auto detection of adapters for paired end data
qualified_quality_phred 30 \- Filters reads based on phred score >=30
unqualified_percent_limit 10 \- percents of bases are allowed to be unqualified, set here as 10%
length_required 100 \- Removes reads shorter than 100 bp. We have read lengths of 150 bp.
cut_right cut_right_window_size 5 cut_right_mean_quality 20- Jill used this sliding cut window in her script. I am going to leave it out for now and evaluate the QC to see if we need to implement cutting.
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=ashuffmyer@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/scripts
#SBATCH -o adapter-trim-%j.out
#SBATCH -e adapter-trim-%j.error
# Load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# Make an array of sequences to trim in raw data directory
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/
array1=($(ls *R1_001.fastq.gz))
echo "Read trimming of adapters started." $(date)
# fastp and fastqc loop
for i in ${array1[@]}; do
fastp --in1 ${i} \
--in2 $(echo ${i}|sed s/_R1/_R2/)\
--out1 /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences/trim.${i} \
--out2 /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences/trim.$(echo ${i}|sed s/_R1/_R2/) \
--detect_adapter_for_pe \
--qualified_quality_phred 30 \
--unqualified_percent_limit 10 \
--length_required 100
done
echo "Read trimming of adapters completed." $(date)
sbatch trim_adapters.sh
Job ID 303864
Ran on Feb 24 2024
An example output from the error file looks like this:
Detecting adapter sequence for read1...
Illumina TruSeq Adapter Read 1: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
Detecting adapter sequence for read2...
No adapter detected for read2
Read1 before filtering:
total reads: 33729404
total bases: 5059410600
Q20 bases: 4895787090(96.766%)
Q30 bases: 4604173440(91.0022%)
Read1 after filtering:
total reads: 21719378
total bases: 3192545540
Q20 bases: 3162771852(99.0674%)
Q30 bases: 3094582490(96.9315%)
Read2 before filtering:
total reads: 33729404
total bases: 5059410600
Q20 bases: 4807178351(95.0146%)
Q30 bases: 4428591566(87.5318%)
Read2 aftering filtering:
total reads: 21719378
total bases: 3192740421
Q20 bases: 3160586478(98.9929%)
Q30 bases: 3088498530(96.735%)
Filtering result:
reads passed filter: 43438756
reads failed due to low quality: 23412708
reads failed due to too many N: 334
reads failed due to too short: 607010
reads with adapter trimmed: 8474961
bases trimmed due to adapters: 213620961
Duplication rate: 62.6758%
Insert size peak (evaluated by paired-end reads): 166
JSON report: fastp.json
HTML report: fastp.html
Completed early morning Feb 25 2024.
Move script out and error files and fastp files to the trimmed sequence folder to keep things organized.
cd raw-sequences
mv adapter-trim* ../trimmed-sequences/
mv fastp* ../trimmed-sequences/
I then downloaded the fastp.html report to look at trimming information.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences/fastp.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC
This file can be found on GitHub here.
Here are the results:
General statistics
| fastp version: | 0.19.7 (https://github.com/OpenGene/fastp) |
|---|---|
| sequencing: | paired end (150 cycles + 150 cycles) |
| mean length before filtering: | 150bp, 150bp |
| mean length after filtering: | 147bp, 147bp |
| duplication rate: | 20.022686% |
| Insert size peak: | 133 |
| Detected read1 adapter: | AGATCGGAAGAGCACACGTCTGAACTCCAGTCA |
Before filtering
| total reads: | 16.841214 M |
|---|---|
| total bases: | 2.526182 G |
| Q20 bases: | 2.421432 G (95.853403%) |
| Q30 bases: | 2.262788 G (89.573449%) |
| GC content: | 44.519841% |
After filtering
| total reads: | 11.487322 M |
|---|---|
| total bases: | 1.692048 G |
| Q20 bases: | 1.678653 G (99.208366%) |
| Q30 bases: | 1.645756 G (97.264174%) |
| GC content: | 43.779533% |
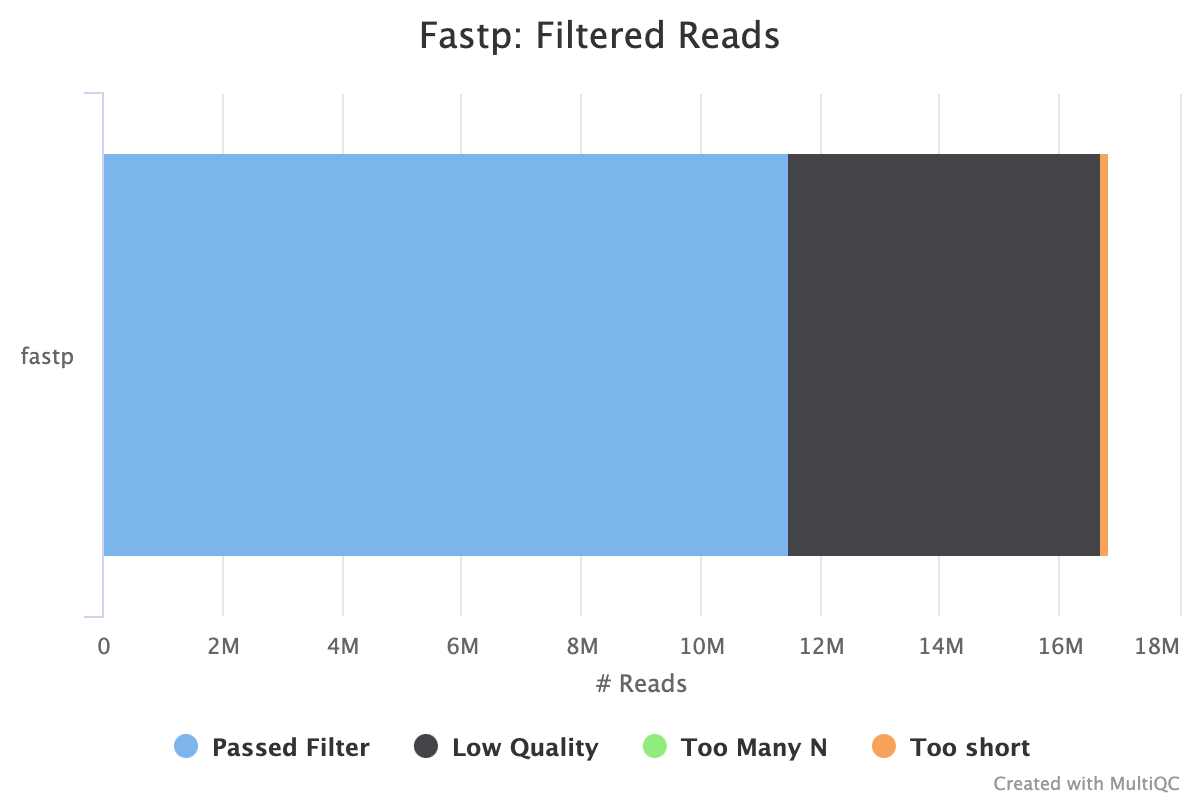
Filtering results
| reads passed filters: | 11.487322 M (68.209584%) |
|---|---|
| reads with low quality: | 5.219646 M (30.993288%) |
| reads with too many N: | 150 (0.000891%) |
| reads too short: | 134.096000 K (0.796237%) |
These results show that filtering improved quality of reads and removed about 30% of reads due to length (a small proportion) and quality (most of reads removed). Average Q30 bases improved from 89% to 97%. Adapters were removed.
Next, I’ll run another round of fastqc and multiqc to see how this changed or improved our qc results.
2. FastQC and MultiQC on trimmed sequences
Make a script for running QC on trimmed sequences.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq
cd scripts
nano qc_trimmed.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=ashuffmyer@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences
#SBATCH -o trimmed-qc-%j.out
#SBATCH -e trimmed-qc-%j.error
# load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# fastqc of raw reads
fastqc *.fastq.gz
#generate multiqc report
multiqc ./ --filename multiqc_report_trimmed.html
echo "Trimmed MultiQC report generated." $(date)
Run the script.
sbatch qc_trimmed.sh
Job 303885 started on 25 Feb in the morning.
Job ended on 25 Feb in the evening.
Make folders for QC files to go into. Move QC files. I tried to set output directories but it was failing, so I’m just manually moving them for now.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences
mkdir trimmed_qc_files
mv *fastqc.html trimmed_qc_files
mv *fastqc.zip trimmed_qc_files
mv multiqc* trimmed_qc_files
mv trimmed-qc-303* trimmed_qc_files
Copy the multiqc html file to my computer.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences/trimmed_qc_files/multiqc_report_trimmed.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC
The MultiQC report results are below. The seven samples that had <10M reads had reads reduced and now they are <5M reads. These will likely need to be removed from analyses. The remaining samples have 9M-25M reads. We will reevaluate which samples should be removed from analyses after the mapping step and after I hear back from Azenta.
This report also included the fastp stats, which was new to me!
Here are some of the main results:
| Sample Name | % Duplication | GC content | % PF | % Adapter | % Dups | % GC | M Seqs |
|---|---|---|---|---|---|---|---|
| fastp | 20.0% | 43.8% | 68.2% | 12.0% | |||
| trim.R100_R1_001 | 75.3% | 43% | 22.6 | ||||
| trim.R100_R2_001 | 75.3% | 43% | 22.6 | ||||
| trim.R101_R1_001 | 75.7% | 43% | 24.7 | ||||
| trim.R101_R2_001 | 75.7% | 43% | 24.7 | ||||
| trim.R102_R1_001 | 75.8% | 43% | 21.7 | ||||
| trim.R102_R2_001 | 75.7% | 43% | 21.7 | ||||
| trim.R103_R1_001 | 73.1% | 43% | 15.0 | ||||
| trim.R103_R2_001 | 73.1% | 43% | 15.0 | ||||
| trim.R104_R1_001 | 72.3% | 43% | 20.3 | ||||
| trim.R104_R2_001 | 72.2% | 43% | 20.3 | ||||
| trim.R105_R1_001 | 72.5% | 43% | 19.3 | ||||
| trim.R105_R2_001 | 72.5% | 43% | 19.3 | ||||
| trim.R106_R1_001 | 72.8% | 43% | 23.3 | ||||
| trim.R106_R2_001 | 72.8% | 43% | 23.3 | ||||
| trim.R107_R1_001 | 50.2% | 43% | 4.8 | ||||
| trim.R107_R2_001 | 50.1% | 43% | 4.8 | ||||
| trim.R108_R1_001 | 72.5% | 43% | 19.6 | ||||
| trim.R108_R2_001 | 72.6% | 43% | 19.6 | ||||
| trim.R55_R1_001 | 72.4% | 43% | 10.4 | ||||
| trim.R55_R2_001 | 72.2% | 43% | 10.4 | ||||
| trim.R56_R1_001 | 71.0% | 44% | 11.0 | ||||
| trim.R56_R2_001 | 70.7% | 44% | 11.0 | ||||
| trim.R57_R1_001 | 72.9% | 43% | 9.8 | ||||
| trim.R57_R2_001 | 72.5% | 43% | 9.8 | ||||
| trim.R58_R1_001 | 72.7% | 43% | 11.6 | ||||
| trim.R58_R2_001 | 72.4% | 43% | 11.6 | ||||
| trim.R59_R1_001 | 39.1% | 43% | 4.2 | ||||
| trim.R59_R2_001 | 38.9% | 43% | 4.2 | ||||
| trim.R60_R1_001 | 71.8% | 43% | 10.4 | ||||
| trim.R60_R2_001 | 71.4% | 43% | 10.4 | ||||
| trim.R61_R1_001 | 73.3% | 43% | 12.8 | ||||
| trim.R61_R2_001 | 73.0% | 43% | 12.8 | ||||
| trim.R62_R1_001 | 72.6% | 43% | 10.6 | ||||
| trim.R62_R2_001 | 72.4% | 43% | 10.6 | ||||
| trim.R63_R1_001 | 72.8% | 43% | 16.4 | ||||
| trim.R63_R2_001 | 72.8% | 43% | 16.4 | ||||
| trim.R64_R1_001 | 72.7% | 43% | 21.6 | ||||
| trim.R64_R2_001 | 72.7% | 43% | 21.6 | ||||
| trim.R65_R1_001 | 72.6% | 43% | 20.9 | ||||
| trim.R65_R2_001 | 72.6% | 43% | 20.9 | ||||
| trim.R66_R1_001 | 74.6% | 43% | 21.7 | ||||
| trim.R66_R2_001 | 74.7% | 43% | 21.7 | ||||
| trim.R67_R1_001 | 52.6% | 42% | 5.4 | ||||
| trim.R67_R2_001 | 52.7% | 43% | 5.4 | ||||
| trim.R68_R1_001 | 72.4% | 43% | 20.7 | ||||
| trim.R68_R2_001 | 72.5% | 43% | 20.7 | ||||
| trim.R69_R1_001 | 74.7% | 44% | 26.1 | ||||
| trim.R69_R2_001 | 74.6% | 44% | 26.1 | ||||
| trim.R70_R1_001 | 73.5% | 43% | 22.7 | ||||
| trim.R70_R2_001 | 73.6% | 43% | 22.7 | ||||
| trim.R71_R1_001 | 71.8% | 43% | 19.0 | ||||
| trim.R71_R2_001 | 71.8% | 43% | 19.0 | ||||
| trim.R72_R1_001 | 72.6% | 43% | 22.0 | ||||
| trim.R72_R2_001 | 72.5% | 43% | 22.0 | ||||
| trim.R73_R1_001 | 73.6% | 43% | 23.2 | ||||
| trim.R73_R2_001 | 73.6% | 43% | 23.2 | ||||
| trim.R74_R1_001 | 72.9% | 44% | 23.4 | ||||
| trim.R74_R2_001 | 73.0% | 44% | 23.4 | ||||
| trim.R75_R1_001 | 55.7% | 43% | 7.0 | ||||
| trim.R75_R2_001 | 55.9% | 43% | 7.0 | ||||
| trim.R76_R1_001 | 74.0% | 43% | 22.1 | ||||
| trim.R76_R2_001 | 74.1% | 43% | 22.1 | ||||
| trim.R77_R1_001 | 73.1% | 43% | 26.7 | ||||
| trim.R77_R2_001 | 73.0% | 43% | 26.7 | ||||
| trim.R78_R1_001 | 73.1% | 43% | 23.2 | ||||
| trim.R78_R2_001 | 73.2% | 43% | 23.2 | ||||
| trim.R79_R1_001 | 73.0% | 44% | 16.7 | ||||
| trim.R79_R2_001 | 72.9% | 44% | 16.7 | ||||
| trim.R80_R1_001 | 73.7% | 43% | 23.5 | ||||
| trim.R80_R2_001 | 73.6% | 43% | 23.5 | ||||
| trim.R81_R1_001 | 71.7% | 44% | 21.5 | ||||
| trim.R81_R2_001 | 71.6% | 44% | 21.5 | ||||
| trim.R82_R1_001 | 74.1% | 43% | 23.4 | ||||
| trim.R82_R2_001 | 74.1% | 43% | 23.4 | ||||
| trim.R83_R1_001 | 51.3% | 43% | 5.3 | ||||
| trim.R83_R2_001 | 51.4% | 43% | 5.3 | ||||
| trim.R84_R1_001 | 74.2% | 43% | 21.3 | ||||
| trim.R84_R2_001 | 74.1% | 43% | 21.3 | ||||
| trim.R85_R1_001 | 74.2% | 43% | 23.8 | ||||
| trim.R85_R2_001 | 74.1% | 43% | 23.8 | ||||
| trim.R86_R1_001 | 73.0% | 43% | 21.0 | ||||
| trim.R86_R2_001 | 73.0% | 43% | 21.0 | ||||
| trim.R87_R1_001 | 71.5% | 43% | 17.0 | ||||
| trim.R87_R2_001 | 71.6% | 43% | 17.0 | ||||
| trim.R88_R1_001 | 73.0% | 44% | 20.0 | ||||
| trim.R88_R2_001 | 73.1% | 44% | 20.0 | ||||
| trim.R89_R1_001 | 73.9% | 43% | 21.8 | ||||
| trim.R89_R2_001 | 74.0% | 43% | 21.8 | ||||
| trim.R90_R1_001 | 73.5% | 43% | 21.6 | ||||
| trim.R90_R2_001 | 73.5% | 43% | 21.6 | ||||
| trim.R91_R1_001 | 51.1% | 43% | 4.8 | ||||
| trim.R91_R2_001 | 51.2% | 43% | 4.8 | ||||
| trim.R92_R1_001 | 76.0% | 43% | 19.0 | ||||
| trim.R92_R2_001 | 76.0% | 43% | 19.0 | ||||
| trim.R93_R1_001 | 83.0% | 45% | 21.3 | ||||
| trim.R93_R2_001 | 83.1% | 45% | 21.3 | ||||
| trim.R94_R1_001 | 76.0% | 43% | 21.3 | ||||
| trim.R94_R2_001 | 76.1% | 43% | 21.3 | ||||
| trim.R95_R1_001 | 73.7% | 43% | 18.3 | ||||
| trim.R95_R2_001 | 73.6% | 43% | 18.3 | ||||
| trim.R96_R1_001 | 74.8% | 44% | 23.0 | ||||
| trim.R96_R2_001 | 74.8% | 44% | 23.0 | ||||
| trim.R97_R1_001 | 75.1% | 43% | 21.5 | ||||
| trim.R97_R2_001 | 75.1% | 43% | 21.5 | ||||
| trim.R98_R1_001 | 73.8% | 43% | 23.5 | ||||
| trim.R98_R2_001 | 73.8% | 43% | 23.5 | ||||
| trim.R99_R1_001 | 54.7% | 43% | 5.7 | ||||
| trim.R99_R2_001 | 54.6% | 43% | 5.7 |
- Fastp filtering: most reads filtered were due to low quality
- Average insert size is 133 bp

- You can see the seven samples here with low read counts (<5M)
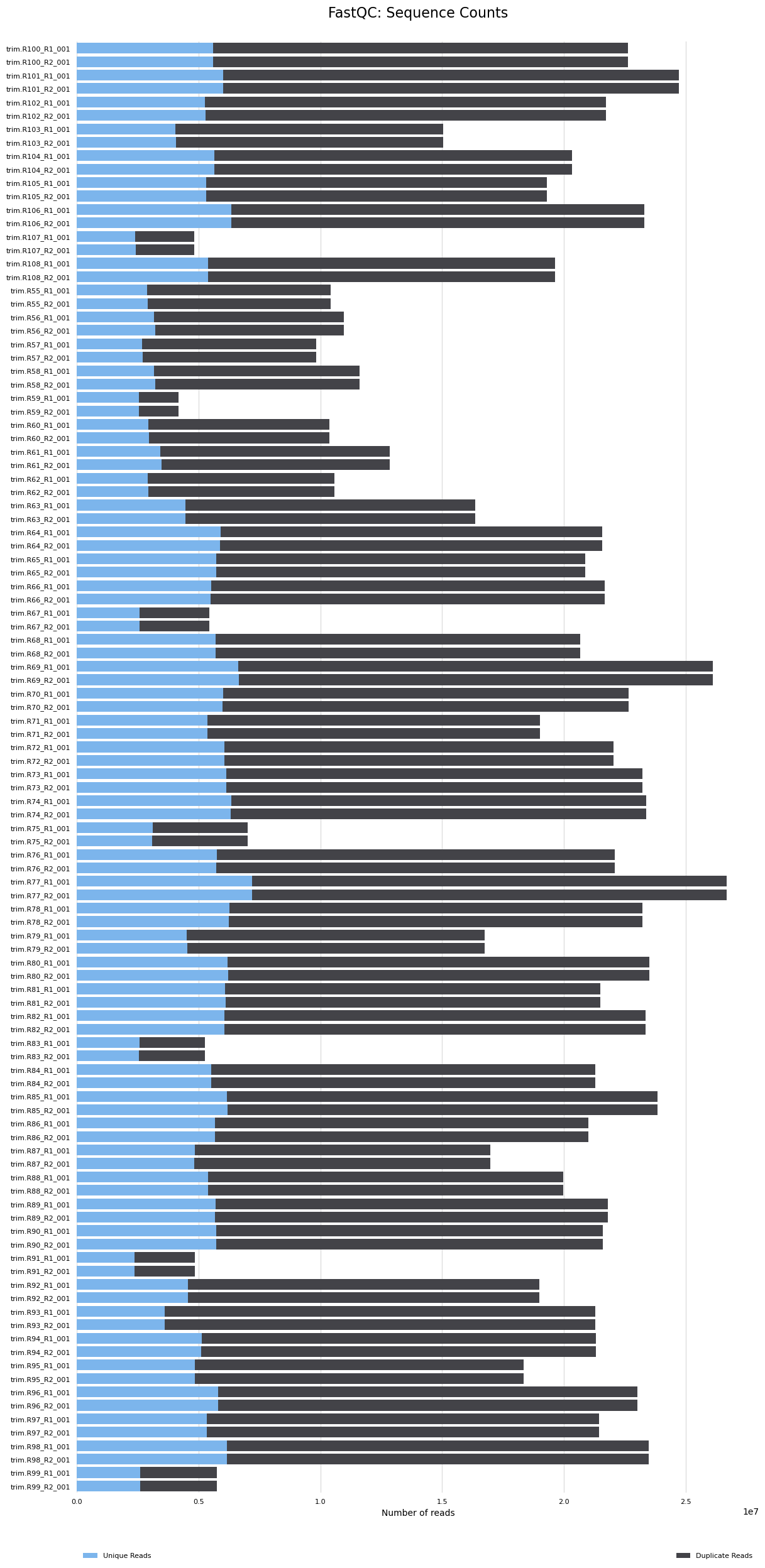
- There is some sequence duplication. Need to determine if this is expected or a problem.

- GC content is better now. There are two samples with weird distributions. I’ll look at those in more detail in individual files.

- Length is shorter now after adapter removal.

- Quality is very high

Next I need to look at the individual raw and trimmed FastQC files for each sample.
Copy the individual fastqc files to my computer.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/raw_fastqc/\*fastqc.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC/fastqc_raw
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences/trimmed_qc_files/\*fastqc.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC/fastqc_trimmed
Raw FastQC files can be found on GitHub here and trimmed FastQC files can be found on GitHub here.
I’ll next dig into each sample and see what they look like - especially those with low sequence counts and deviations from normal QC results.