


QC of top off RNAseq files for Montipora capitata larval thermal tolerance 2023 project
This post details QC of the Montipora capitata 2023 larval thermal tolerance project RNAseq files from second delivery of top off sequencing. See my notebook posts and my GitHub repo for information on this project.
Also see my notebook post on sequence download and SRA upload for these files.
1. Run MultiQC on raw data from second sequencing (untrimmed and unfiltered)
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/scripts
nano qc_raw_second_sequencing.sh
#!/bin/bash
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=20
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --output="qc_raw_second_seq-%j.out"
#SBATCH --error="qc_raw_second_seq-%j.err"
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/second_sequencing
# load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# fastqc of raw reads
fastqc *.fastq.gz
#generate multiqc report
multiqc ./ --filename multiqc_report_raw_second_sequencing.html
echo "Raw MultiQC report generated." $(date)
Run the script.
sbatch qc_raw_second_sequencing.sh
Move .md5 files to md5_files folder.
mv ./*.md5 md5_files
Run as Job ID 312074 on 13 April 2024
Move .fastqc files to a QC folder to keep folder organized within the second_sequencing directory.
mkdir raw_fastqc
mv *fastqc.html raw_fastqc
mv *fastqc.zip raw_fastqc
mv multiqc* raw_fastqc
Download the multiQC report to my desktop to view (in a new terminal not logged into Andromeda).
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/second_sequencing/raw_fastqc/multiqc_report_raw_second_sequencing.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC
Move individual fastqc files to local machine. fastqc_second_sequencing_raw
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/second_sequencing/raw_fastqc/\*fastqc.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC/fastqc_second_sequencing_raw
The raw sequence MultiQC report can be found on GitHub here.
Here are the things I noticed from the QC report:
See this previous post with statistics of samples provided by Azenta.
- Top off sequencing
- Samples that had the lowest read depth in the first round of sequencing have the highest read depth in this top off sequencing. Azenta sequenced samples to bring all samples to a total of ~18M reads. This is why there are variable read depths in these top off sequences.
| Sample Name | % Dups | % GC | M Seqs |
|---|---|---|---|
| R107s_R1_001 | 53.2% | 43% | 17.5 |
| R107s_R2_001 | 50.7% | 44% | 17.5 |
| R55s_R1_001 | 34.0% | 43% | 2.9 |
| R55s_R2_001 | 32.5% | 44% | 2.9 |
| R56s_R1_001 | 29.5% | 44% | 1.7 |
| R56s_R2_001 | 27.9% | 45% | 1.7 |
| R57s_R1_001 | 37.2% | 43% | 3.3 |
| R57s_R2_001 | 35.4% | 44% | 3.3 |
| R58s_R1_001 | 30.8% | 43% | 1.4 |
| R58s_R2_001 | 29.1% | 44% | 1.4 |
| R59s_R1_001 | 56.8% | 44% | 33.7 |
| R59s_R2_001 | 54.6% | 44% | 33.7 |
| R60s_R1_001 | 33.0% | 44% | 2.5 |
| R60s_R2_001 | 31.5% | 44% | 2.5 |
| R62s_R1_001 | 34.7% | 43% | 2.3 |
| R62s_R2_001 | 32.9% | 44% | 2.3 |
| R67s_R1_001 | 48.2% | 43% | 17.4 |
| R67s_R2_001 | 46.2% | 44% | 17.4 |
| R75s_R1_001 | 46.0% | 43% | 11.1 |
| R75s_R2_001 | 44.2% | 44% | 11.1 |
| R83s_R1_001 | 52.9% | 43% | 17.6 |
| R83s_R2_001 | 49.4% | 44% | 17.6 |
| R91s_R1_001 | 54.1% | 43% | 17.5 |
| R91s_R2_001 | 52.5% | 44% | 17.5 |
| R99s_R1_001 | 51.1% | 44% | 16.2 |
| R99s_R2_001 | 46.8% | 44% | 16.2 |
Statistics are similar to the values from the original sequencing for these samples.
- Adapter content present in sequences, as expected, because we have not yet removed adapters.

- Some samples have warnings for GC content. We will revisit this after trimming.
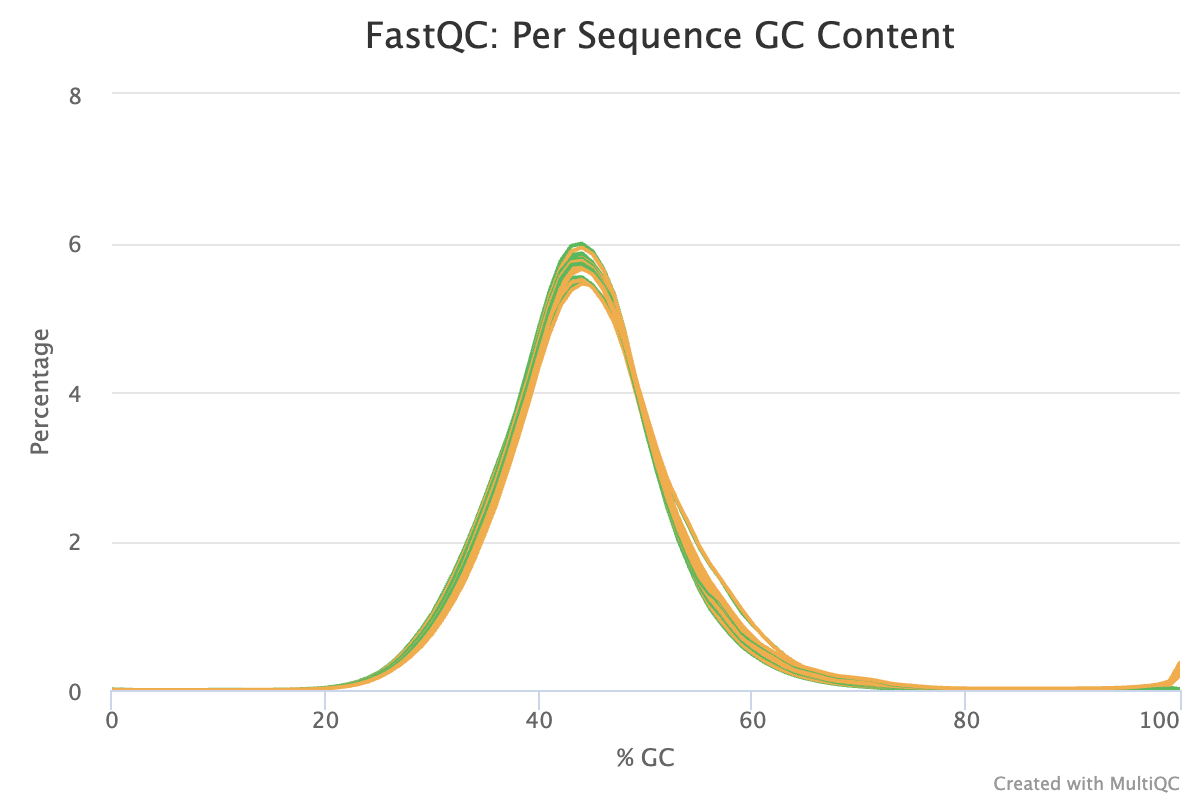
- There are a high proportion of overrepresented sequences. This is expected with RNAseq data as there are likely genes that are highly and consistently expressed.

-
All reads are the same length (150 bp).
-
High quality scores.
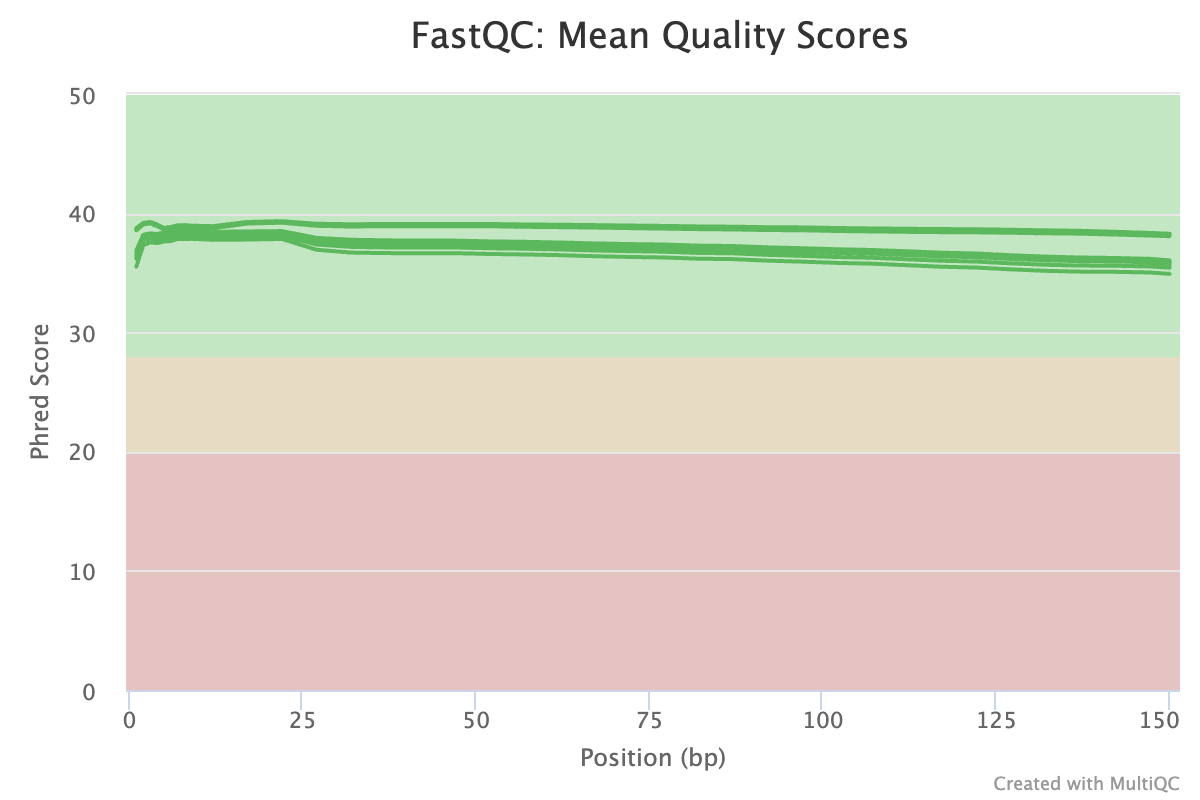
- Low N content

2. Trimming adapters
I ran trimming steps using the script from trimming original sequences, detailed in my previous post.
I first moved the .md5 files to an md5 folder to keep only .fastq files in raw-sequences.
Make a new folder for trimmed sequences.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/
mkdir trimmed-sequences-second-seq
cd scripts
nano trim_adapters_second_seq.sh
I will use the following settings for trimming in fastp. Fastp documentation can be found here.
detect_adapter_for_pe \- This enables auto detection of adapters for paired end data
qualified_quality_phred 30 \- Filters reads based on phred score >=30
unqualified_percent_limit 10 \- percents of bases are allowed to be unqualified, set here as 10%
length_required 100 \- Removes small reads shorter than 100 bp. We have read lengths of 150 bp.
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=ashuffmyer@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/scripts
#SBATCH -o adapter-trim-second-seq-%j.out
#SBATCH -e adapter-trim-second-seq-%j.error
# Load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# Make an array of sequences to trim in raw data directory
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/raw-sequences/second_sequencing
array1=($(ls *R1_001.fastq.gz))
echo "Read trimming of adapters started." $(date)
# fastp and fastqc loop
for i in ${array1[@]}; do
fastp --in1 ${i} \
--in2 $(echo ${i}|sed s/_R1/_R2/)\
--out1 /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq/trim.${i} \
--out2 /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq/trim.$(echo ${i}|sed s/_R1/_R2/) \
--detect_adapter_for_pe \
--qualified_quality_phred 30 \
--unqualified_percent_limit 10 \
--length_required 100
done
echo "Read trimming of adapters completed." $(date)
sbatch trim_adapters_second_seq.sh
Job ID 312075
Ran on April 13 2024, completed in about 1.5 hours.
An example output from the error file looks like this:
Detecting adapter sequence for read1...
Illumina TruSeq Adapter Read 1: AGATCGGAAGAGCACACGTCTGAACTCCAGTCA
Detecting adapter sequence for read2...
No adapter detected for read2
Read1 before filtering:
total reads: 17533977
total bases: 2630096550
Q20 bases: 2576383048(97.9577%)
Q30 bases: 2477902823(94.2134%)
Read1 after filtering:
total reads: 11457851
total bases: 1693380571
Q20 bases: 1685110826(99.5116%)
Q30 bases: 1663210624(98.2184%)
Read2 before filtering:
total reads: 17533977
total bases: 2630096550
Q20 bases: 2486634287(94.5454%)
Q30 bases: 2272601196(86.4075%)
Read2 aftering filtering:
total reads: 11457851
total bases: 1693487809
Q20 bases: 1677451996(99.0531%)
Q30 bases: 1639980332(96.8404%)
Filtering result:
reads passed filter: 22915702
reads failed due to low quality: 11923964
reads failed due to too many N: 146
reads failed due to too short: 228142
reads with adapter trimmed: 3211274
bases trimmed due to adapters: 77884301
Duplication rate: 34.8162%
Insert size peak (evaluated by paired-end reads): 186
JSON report: fastp.json
HTML report: fastp.html
Move script out and error files and fastp files to the trimmed sequence folder to keep things organized.
cd raw-sequences/second_sequencing
mv fastp* ../../trimmed-sequences-second-seq/
cd ../../trimmed-sequences-second-seq/
mv fastp.html fastp_second_seq.html
I then downloaded the fastp.html report to look at trimming information.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq/fastp_second_seq.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC/
This file can be found on GitHub here.
Here are the results:
General statistics
| fastp version: | 0.19.7 (https://github.com/OpenGene/fastp) |
|---|---|
| sequencing: | paired end (150 cycles + 150 cycles) |
| mean length before filtering: | 150bp, 150bp |
| mean length after filtering: | 147bp, 147bp |
| duplication rate: | 30.411524% |
| Insert size peak: | 169 |
| Detected read1 adapter: | AGATCGGAAGAGCACACGTCTGAACTCCAGTCA |
Before filtering
| total reads: | 32.443162 M |
|---|---|
| total bases: | 4.866474 G |
| Q20 bases: | 4.629020 G (95.120605%) |
| Q30 bases: | 4.274311 G (87.831783%) |
| GC content: | 44.578400% |
After filtering
| total reads: | 18.461102 M |
|---|---|
| total bases: | 2.725187 G |
| Q20 bases: | 2.703615 G (99.208398%) |
| Q30 bases: | 2.651870 G (97.309640%) |
| GC content: | 43.742790% |
Filtering results
| reads passed filters: | 18.461102 M (56.902906%) |
|---|---|
| reads with low quality: | 13.784328 M (42.487622%) |
| reads with too many N: | 72 (0.000222%) |
| reads too short: | 197.660000 K (0.609250%) |
These results show that filtering improved quality of reads and removed about 45% of reads due to length (a small proportion) and quality (most of reads removed). Average Q30 bases improved from 87% to 97%. Adapters were removed.
Next, I’ll run another round of fastqc and multiqc to see how this changed or improved our qc results.
3. FastQC and MultiQC on trimmed sequences
Make a script for running QC on trimmed sequences.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/scripts
nano qc_trimmed_second_seq.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=ashuffmyer@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq
#SBATCH -o trimmed-qc-second-seq-%j.out
#SBATCH -e trimmed-qc-second-seq-%j.error
# load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# fastqc of raw reads
fastqc *.fastq.gz
#generate multiqc report
multiqc ./ --filename multiqc_report_trimmed_second_seq.html
echo "Trimmed MultiQC report generated." $(date)
Run the script.
sbatch qc_trimmed_second_seq.sh
Job 312079 started on 13 April, ended on 13 April.
Make folders for QC files to go into.
cd /data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq
mkdir trimmed_qc_files
mv *fastqc.html trimmed_qc_files
mv *fastqc.zip trimmed_qc_files
mv multiqc* trimmed_qc_files
Copy the multiqc html file to my computer.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq/trimmed_qc_files/multiqc_report_trimmed_second_seq.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC
Here are some of the main results:
| Sample Name | % Duplication | GC content | % PF | % Adapter | % Dups | % GC | M Seqs |
|---|---|---|---|---|---|---|---|
| fastp | 30.4% | 43.7% | 56.9% | 8.9% | |||
| trim.R107s_R1_001 | 51.3% | 43% | 11.5 | ||||
| trim.R107s_R2_001 | 51.1% | 43% | 11.5 | ||||
| trim.R55s_R1_001 | 32.7% | 43% | 1.9 | ||||
| trim.R55s_R2_001 | 32.6% | 43% | 1.9 | ||||
| trim.R56s_R1_001 | 28.0% | 44% | 1.1 | ||||
| trim.R56s_R2_001 | 27.8% | 44% | 1.1 | ||||
| trim.R57s_R1_001 | 35.7% | 43% | 2.2 | ||||
| trim.R57s_R2_001 | 35.6% | 43% | 2.2 | ||||
| trim.R58s_R1_001 | 29.5% | 43% | 0.9 | ||||
| trim.R58s_R2_001 | 29.3% | 43% | 0.9 | ||||
| trim.R59s_R1_001 | 54.8% | 43% | 22.1 | ||||
| trim.R59s_R2_001 | 54.9% | 43% | 22.1 | ||||
| trim.R60s_R1_001 | 32.0% | 43% | 1.7 | ||||
| trim.R60s_R2_001 | 31.9% | 43% | 1.7 | ||||
| trim.R62s_R1_001 | 33.3% | 43% | 1.5 | ||||
| trim.R62s_R2_001 | 33.2% | 43% | 1.5 | ||||
| trim.R67s_R1_001 | 46.2% | 43% | 11.3 | ||||
| trim.R67s_R2_001 | 46.3% | 43% | 11.3 | ||||
| trim.R75s_R1_001 | 44.1% | 43% | 7.3 | ||||
| trim.R75s_R2_001 | 44.2% | 43% | 7.3 | ||||
| trim.R83s_R1_001 | 50.0% | 43% | 10.7 | ||||
| trim.R83s_R2_001 | 49.7% | 43% | 10.7 | ||||
| trim.R91s_R1_001 | 52.5% | 43% | 11.6 | ||||
| trim.R91s_R2_001 | 52.9% | 43% | 11.6 | ||||
| trim.R99s_R1_001 | 48.2% | 43% | 9.2 | ||||
| trim.R99s_R2_001 | 47.7% | 43% | 9.2 |
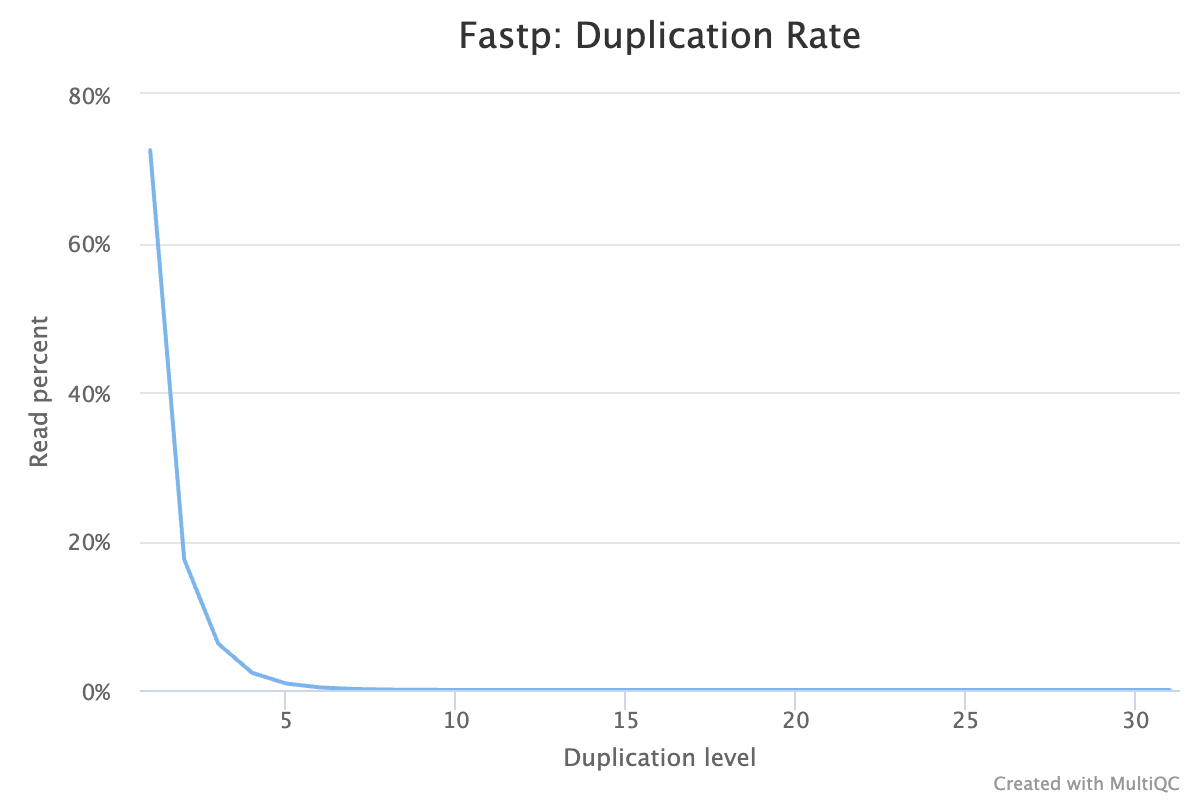
- Fastp filtering: most reads filtered were due to low quality

- Average insert size is 133 bp and within expected range (150 with total overlap to <300 with less overlap).
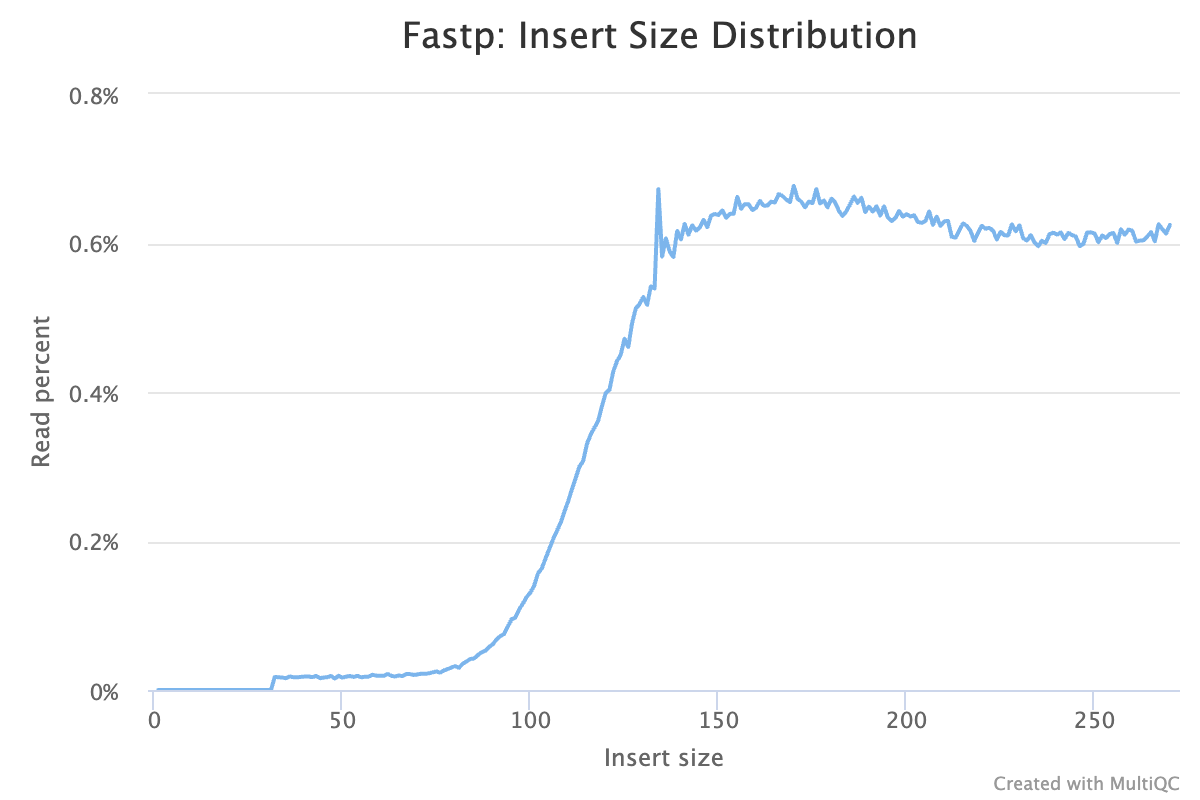
- Read depth is highest for samples with lowest read depth in first round of sequencing.

- Duplication passes QC.

- GC content passes QC.

- Quality is very high
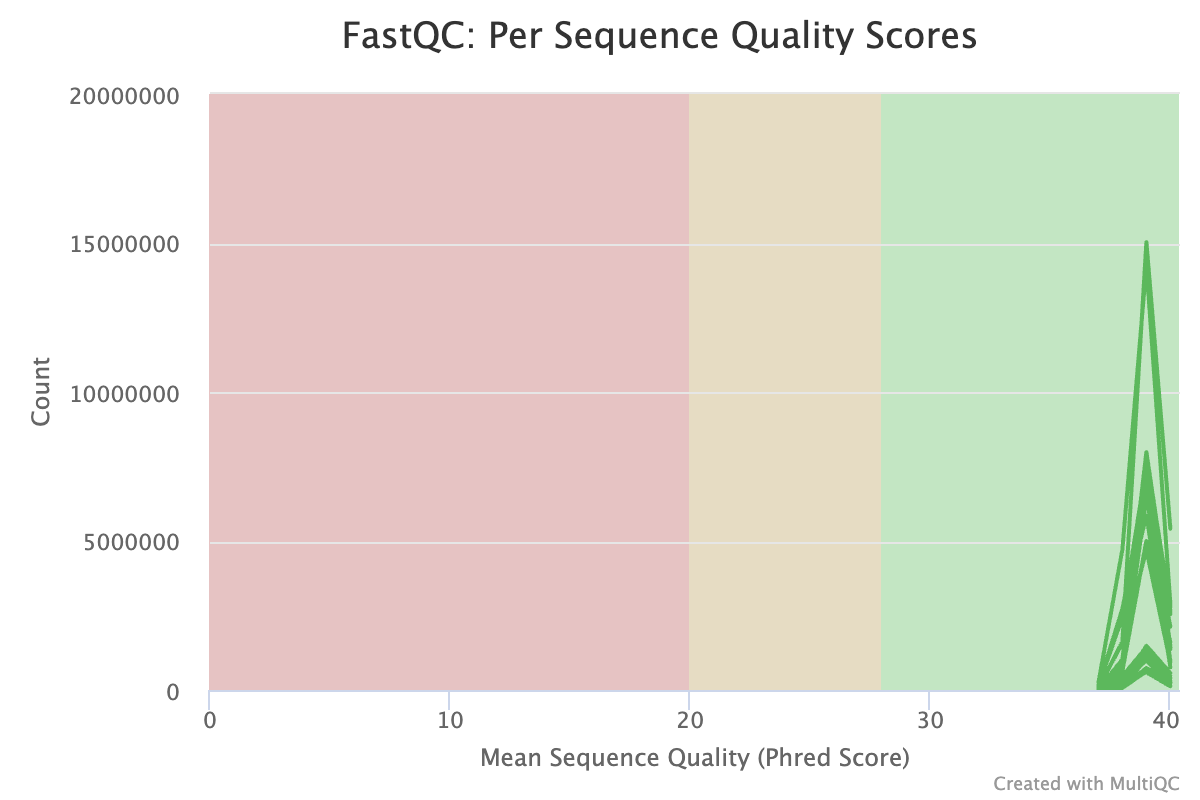

- Low N
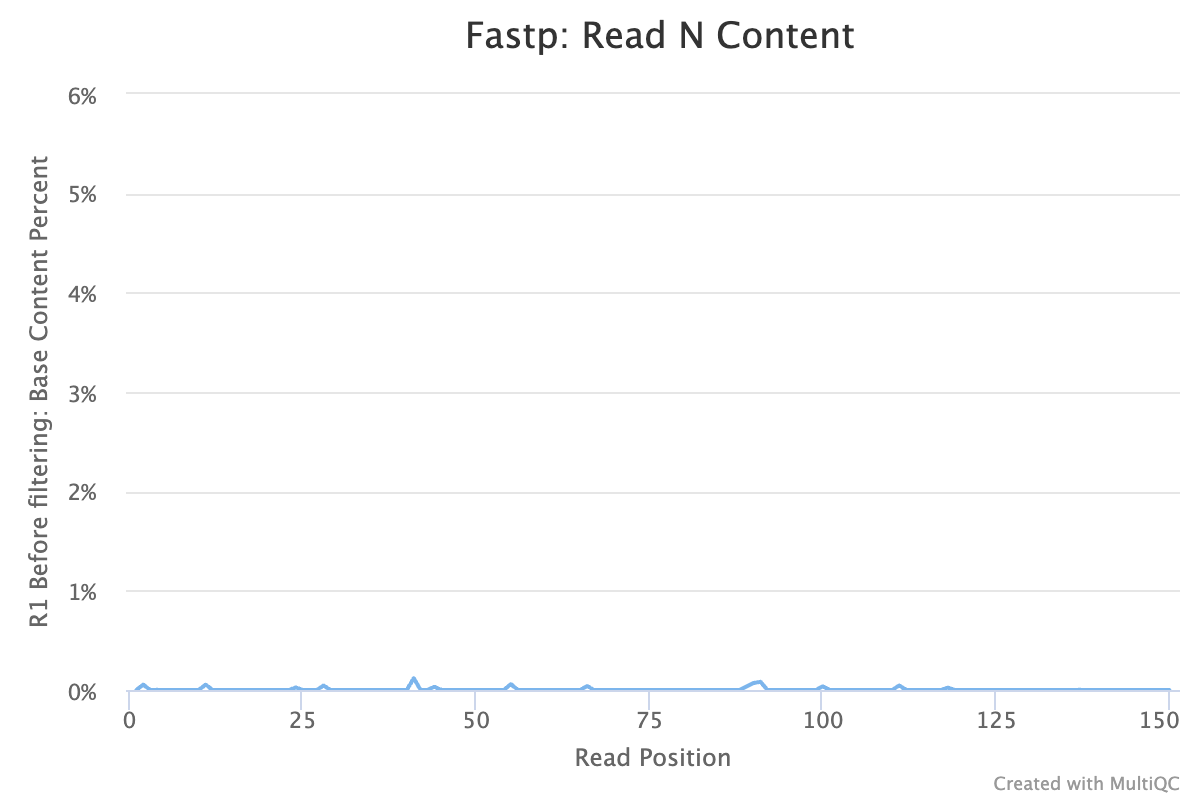
No samples found with any adapter contamination > 0.1%.
Samples had less than 1% of reads made up of overrepresented sequences.
Copy the individual fastqc files to my computer.
scp ashuffmyer@ssh3.hac.uri.edu:/data/putnamlab/ashuffmyer/mcap-2023-rnaseq/trimmed-sequences-second-seq/trimmed_qc_files/\*fastqc.html ~/MyProjects/larval_symbiont_TPC/data/rna_seq/QC/fastqc_second_sequencing_trimmed
Raw FastQC files can be found on GitHub here and trimmed FastQC files can be found on GitHub here.